
Error loading player: No playable sources found
3592600
Progress in protein-ligand binding free energy calculations
Date
August 23, 2021
The mining minima algorithm for computing binding free energies has provided good correlations with experiment for a range of protein-ligand systems, along with enhanced computational throughput. It uses conformational search to discover the chief energy minima, computes the configurational integral in each energy well, and sums over minima to yield an overall free energy. Doing this for the bound and free states provides the change in free energy on binding. The latest VM2 implementation of mining minima is automated, uses GPU acceleration, and supports both empirical force fields and quantum mechanical methods. Recent advances, including steps toward accessing VM2 through the Molecular Operating Environment (MOE) graphical user interface, will be described.
Computationally intensive free energy methods based on molecular dynamics (MD) simulations with explicit solvent are also of high interest, but can become problematic when the protein’s binding site is buried within the protein. In such systems, changes in hydration of the buried ligand that ought to occur during alchemical changes or decoupling of the ligand may not, because it takes too long for the site to sample configurations open to bulk solvent. Adding large Monte Carlo translational water moves to traditional MD can overcome this problem, improving convergence and accuracy of relative binding free energy calculations. Recently, bottleneck steps in this hybrid MD/MD method have been ported to GPU, greatly accelerating the calculations.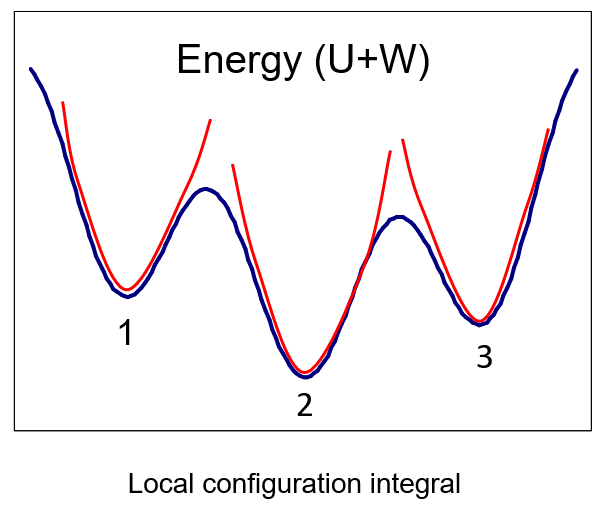
Computationally intensive free energy methods based on molecular dynamics (MD) simulations with explicit solvent are also of high interest, but can become problematic when the protein’s binding site is buried within the protein. In such systems, changes in hydration of the buried ligand that ought to occur during alchemical changes or decoupling of the ligand may not, because it takes too long for the site to sample configurations open to bulk solvent. Adding large Monte Carlo translational water moves to traditional MD can overcome this problem, improving convergence and accuracy of relative binding free energy calculations. Recently, bottleneck steps in this hybrid MD/MD method have been ported to GPU, greatly accelerating the calculations.
The mining minima algorithm estimates overall free energy in terms of a sum over energy wells.
Presenter

Professor, U. C. San Diego
Speakers
Iowa State University
University of Colorado Denver
Related Products

Effective fragment molecular orbital (EFMO): Nanomaterial, biomolecules, and beyond
Accurately and efficiently modeling nano-sized molecular clusters and beyond can now be achieved with an _ab initio-_based fragmentation approach, the effective fragment molecular orbital (EFMO) method…
An interface of statistical mechanics with quantum potentials for host-guest binding affinity calculations
To improve accuracy for calculations of host-guest binding properties, Verachem’s classical force-field-based free energy prediction (VM2) methods have been integrated with quantum energy potentials…
Effective fragment molecular orbital (EFMO): Nanomaterial, biomolecules, and beyond
Accurately and efficiently modeling nano-sized molecular clusters and beyond can now be achieved with an _ab initio-_based fragmentation approach, the effective fragment molecular orbital (EFMO) method…
An interface of statistical mechanics with quantum potentials for host-guest binding affinity calculations
To improve accuracy for calculations of host-guest binding properties, Verachem’s classical force-field-based free energy prediction (VM2) methods have been integrated with quantum energy potentials…
